The FDA just approved one of the first drugs for ALS. Some raise questions, but this Penn patient has no doubt.
Neurologist Toby Ferguson, formerly of Temple University and Penn Medicine, helped develop the Biogen drug, to be sold as Qalsody.
Todd Legg started to have trouble breathing in December 2019. Around the same time, he noticed that his hands and arms cramped up when washing dishes at home in Brackney, Pa., north of Scranton.
His wife feared the worst, remembering that Legg’s mother had died of the nerve-wasting disease called amyotrophic lateral sclerosis (ALS), which causes loss of muscle control and eventually robs people of their ability to walk, talk, eat, and breathe.
No way, Legg said. He was only 47. It was wintertime, so he was probably just out of shape or dehydrated, he reasoned.
Yet within months, after his breathing grew weaker and weaker, Legg’s doctors determined that his wife’s hunch was correct. He had ALS, and he likely would become paralyzed and die within a few years. The doctors said there was little they could do — unless he was willing to try an experimental new drug.
Two years after Legg started getting injections of the drug at the University of Pennsylvania, his breathing ability has remained fairly stable, good enough for him to hunt, fish, and coach his son’s Little League baseball team. On Tuesday, the FDA granted accelerated approval to the drug, tofersen, meaning others with his type of genetic mutation will be able to get the treatment, too — if insurers agree to pay.
The news was cheered by people with the deadly disease, even though most of them have forms of the disease for which the drug will not work. Most cases have no known genetic cause. About 10% of cases are caused by a genetic mutation, and tofersen is designed to treat a fraction of those. And even for those who do qualify for the treatment, there is debate over how effective it is.
In the first six months of a clinical trial, patients getting the drug fared no better than those who received a placebo, as measured on a combined scale of everyday functions such as breathing, swallowing, and walking. Still, those who received the drug had significantly lower levels of a key biomarker — a telltale substance in the blood that indicates when the person’s nerves are degenerating.
That was enough for FDA to grant accelerated approval, a pathway reserved for serious diseases without good treatments. Further research is underway to confirm that the drug, to be sold as Qalsody, is effective, including a study of people with the mutation who have yet to develop symptoms.
In Legg’s case, Penn Medicine neurologist Colin Quinn has no doubt that the drug worked.
Yet he almost didn’t get to try it. When the research team first evaluated Legg, his breathing ability had declined too much for him to qualify for the trial.
So the team sent a nurse to Legg’s house in Susquehanna County to measure his breathing strength a second time, and he just made it. Within days, in October 2020, he was in Philadelphia to get the first injection.
“That’s the difference between him being alive today and not,” Quinn said. “Which is so scary to think about. I actually try not to think about it.”
How the ALS drug works
In patients with Legg’s form of ALS, the genetic mutation causes them to make an abnormal, toxic version of a protein called SOD1. As the protein accumulates in the central nervous system, it causes deterioration in the nerves that control muscles, which start to wither away as a result. Patients — currently numbering several hundred in the U.S. — typically die within a few years.
Tofersen is designed to work by tamping down the body’s ability to make the toxic protein. It consists of short strands of genetic material called antisense oligonucleotides (ASO), delivered via periodic injections into the lower spine.
It is made by Cambridge, Mass.-based Biogen. Among the scientists overseeing the drug’s development at the company is Toby Ferguson, formerly a researcher at Temple University and Penn.
After a few months on the drug, levels of the protein decline by about one-third — enough to dramatically slow the rate of nerve damage, Quinn says.
Legg, a high school math teacher who came to Penn on Tuesday for his 36th injection, likes to repeat an analogy that an acquaintance came up with:
“Let’s say you have a pond and it’s full of some kind of fish or critter that you want out, so you put in there a bigger fish that’s going to eat them,” he said. “It’s not going to happen overnight.”
Legg felt strongly enough about the drug’s impact that on March 22, he spoke before an FDA advisory committee that was reviewing the evidence for the drug.
He described his August 2020 diagnosis as a “death sentence.” He realized that he must have inherited the fatal mutation from his mother, though she was never tested before dying in 2010.
“I was 47 years old,” he told the panel, speaking via teleconference. “I wasn’t sure if I was going to make it to 50. And here I am.”
The evidence
FDA’s process for approving certain drugs without conclusive evidence has sparked controversy.
The agency followed the same pathway in its recent approval of a drug for Alzheimer’s, called Leqembi. As with the drug for ALS, the strongest evidence in favor of Leqembi consisted of biomarkers — indirect measurements suggesting that the drug is effective.
Underwhelmed by the results, the Centers for Medicare and Medicaid Services has declined to pay for the drug until additional evidence emerges. Yet in March, the Veterans Health Administration agreed to cover the drug for veterans.
Quinn, the Penn neurologist, is optimistic that there will be no such uncertainty with the ALS drug — that all insurers will cover it.
He cited the fact that after the first six months of the trial, participants who had been taking the placebo switched to getting the real drug. And in the two years since then, their condition appears to have stabilized somewhat.
David Weisman, an Abington-based neurologist who served on the FDA advisory panel that reviewed the evidence for the ALS drug, agreed that signs are promising.
“I think it works over a longer time frame,” he said.
On March 22, Weisman and his fellow advisers voted unanimously that the drug was “reasonably likely” to prove beneficial, paving the way for the agency to approve it Tuesday.
The injections
That happened to be the same day as Legg’s 36th injection.
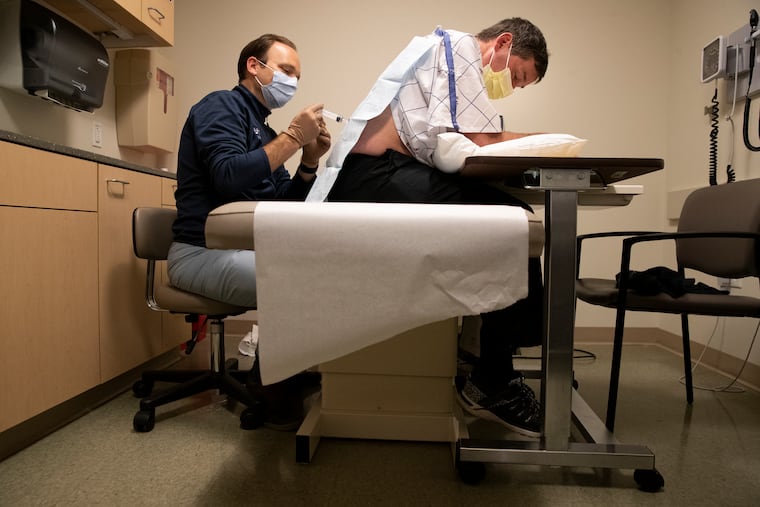
Sitting on an examination table at Penn’s Perelman Center for Advanced Medicine, Legg leaned forward so that Quinn could lift the back of his hospital gown and insert a slender needle into his lower spine.
First, the physician drew a few drops of Legg’s spinal fluid for analysis. Then he injected the drug, slowly depressing a syringe over the course of two minutes, as research coordinator Adreeja GuhaRay called out the time.
The first three injections in 2021 were spaced two weeks apart. Since then, they’ve been every four weeks.
By now, Legg is so comfortable with the process that even with a needle in his lower back, he is able to swipe through pictures on his smartphone.
At home in Brackney, he has learned to live with his condition. Though the drug appears to have slowed his nerve damage and stabilized his breathing ability, the muscles in his right arm have grown substantially weaker.
So as a natural right-hander, the math teacher taught himself to write left-handed on the board in his classroom. Last week, he even managed to throw left-handed for his son’s Little League team to have batting practice.
“I keep trying to everything that I can do,” he said. “It’s not nearly as fast. Maybe not as good. But we try to do what we can.”
